Notre mission
Faire progresser la recherche en sciences de la vie grâce à des technologies peptidiques innovantes et à un service client exceptionnel.
Notre Vision
Devenir un leader mondialement reconnu dans le domaine de la synthèse peptidique et des domaines biotechnologiques connexes.

Dans le suc gastrique humain, le BPC 157 est stable pendant plus de 24 heures et présente donc une bonne biodisponibilité orale (toujours administré seul) et des effets bénéfiques sur l'ensemble du tractus gastro-intestinal. Il s'agit d'une distinction importante par rapport aux autres peptides standards, qui dépendent fonctionnellement de l'ajout d'un support ou sont rapidement détruits dans le suc gastrique humain. Par conséquent, il est suggéré que le BPC 157 stable soit un médiateur de la cytoprotection de Robert, qui maintient l'intégrité de la muqueuse ogastro-intestinale. Nous suggérons que la contribution du BPC 157 à la cytoprotection de Robert – c’est-à-dire la capacité à contrecarrer les lésions gastriques fondamentales induites par l’alcool, que Robert a appelées cytoprotection – et la capacité à contrecarrer les lésions résultant du contact nocif direct de l’agent nocif avec la cellule représentent la connexion périphérique entre l’intestin et l’axe cérébral.
Perovic a rapporté que le BPC 157 avait un effet thérapeutique marqué sur la guérison des rats présentant une lésion de la moelle épinière avec paralysie de la queue (lésion par compression d'une minute de la moelle épinière sacrocaudale [S2 – Co1]). Plus précisément, une seule administration intrapéritonéale de BPC 157 10 minutes après la blessure neutralise les effets négatifs. En revanche, les lésions de la moelle épinière et la paralysie de la queue persistent chez les rats non traités, évaluées des jours, des semaines, des mois et un an après la blessure. Il convient de noter que le BPC 157 atténue les dommages généralement causés. Ainsi, la thérapie BPC 157 entraîne une récupération fonctionnelle, microscopique et électrophysiologique évidente.



Il convient de noter que chez les rats présentant une lésion de la moelle épinière, il existe une reperfusion permanente. Une fois que le BPC 157 est administré 10 minutes après une lésion de compression, il existe une protection continue et aucune perturbation spontanée induite par une lésion de la moelle épinière ne réapparaît. Toutes les lésions de la moelle épinière provoquent immédiatement une hémorragie, avec la mort ultérieure des neurones et des oligodendrocytes.
Il est donc concevable qu’une hémostase précoce puisse être bénéfique et permettre une récupération fonctionnelle après une contusion médullaire chez le rat. Cependant, l’effet exercé par le BPC 157 est probablement différent du simple effet hémostatique qui atténuerait les lésions de la moelle épinière, car le BPC 157 améliore également nettement la fonction thrombocytaire chez le rat sans affecter les facteurs de coagulation. Pendant la récupération d'une lésion médullaire, le BPC 157 protège également directement l'endothélium, atténue les troubles de l'occlusion vasculaire périphérique, active rapidement des voies de contournement alternatives et neutralise les syndromes induits par l'occlusion veineuse. Ainsi, en supposant qu'il existe une contribution veineuse substantielle à la compression de la moelle épinière, il est concevable que le flux sanguin rétabli médié par le BPC 157 puisse sans aucun doute contribuer à l'effet de récupération rapide. De plus, étant donné que le BPC 157 favorise la reperfusion permanente après compression de la moelle épinière, il convient de noter que lorsque le BPC 157 est administré pendant la reperfusion, il neutralise les accidents vasculaires cérébraux induits par le clampage bilatéral des artères carotides communes. BPC 157 résout les dommages neuronaux et prévient les déficits de mémoire, de locomotion et de coordination. BPC 157 exerce apparemment ces effets en modifiant l'expression des gènes dans l'hippocampe.
En conclusion, le BPC 157 exerce des effets bénéfiques sur les accidents vasculaires cérébraux, la schizophrénie et les lésions de la moelle épinière.
Les chercheurs ont constamment démontré que le BPC 157 exerce une myriade d’effets bénéfiques dans tout le corps. Il n'y a aucune raison d'indiquer que les avantages du BPC 157 soient limités par la validité des modèles utilisés et/ou par les limites de la méthodologie. En effet, nous pouvons affirmer que l’efficacité, la facilité d’application, le profil clinique sûr et le mécanisme du BPC 157 représentent une orientation thérapeutique alternative, probablement réussie, pour les affections neurologiques. Par conséquent, des études supplémentaires sont nécessaires pour clarifier comment une thérapie potentielle par BPC 157 traiterait spécifiquement un mécanisme d'action impliquant plusieurs sites subcellulaires du SNC. L’influence sur le fonctionnement de la plupart, sinon de la totalité, des systèmes neuronaux aux niveaux moléculaire, cellulaire et systémique devrait être explorée. Certains relais viscéraux répétitifs du SNC ou des organes circumventriculaires, l'une des rares régions du cerveau sans barrière hémato-encéphalique, constituent une voie connue par laquelle un peptide administré par voie systémique peut exercer un effet central. Ainsi, il doit agir dans l’axe intestin-cerveau, que cette action soit directe ou indirecte.
La peptstatine est un pentapeptide, un inhibiteur naturel de l'aspartyl protéase, qui peut inhiber la protéase aspartique et la protéase acide de divers micro-organismes. La peptstatine est principalement sécrétée par les espèces Streptomyces et produite par Streptomyces. Il peut inhiber la pepsine, la pepsine D et l'enzyme libérant l'angiotensine, et a des effets thérapeutiques sur l'ulcère gastrique, l'hypertension rénale, l'arthrite, l'œdème carraghénane et d'autres maladies.
La pepstine est un puissant inhibiteur des aspartyl protéases telles que la pepsine, la cathepsine D et la rénine. Ce pentapeptide naturel isolé des actinomycètes a été pendant de nombreuses années l'inhibiteur classique de la rénine in vitro. La pepstatine n'est pas spécifique de la rénine et est peu soluble dans l'eau. Les dérivés structurels de la pepstatine ont augmenté sa solubilité et sa spécificité pour la rénine de plusieurs ordres de grandeur. La pepstatine contient une statine d'acide aminé γ inhabituelle qui peut remplacer les deux acides aminés au niveau de la liaison scissile du substrat protéique et bloquer le clivage du substrat en raison de l'analogie structurelle avec un état de transition de l'hydrolyse de la liaison peptidique par les aspartyl protéases.
Lorsque la protéine est extraite de cellules brisées, des protéases peuvent être libérées, qui doivent être rapidement inhibées pour empêcher la dégradation des protéines. Lors du processus d’extraction des protéines, des inhibiteurs de protéase doivent être ajoutés pour empêcher la protéolyse. L'inhibiteur de protéase fait généralement référence à une substance qui se lie à certains groupes du centre actif des molécules de protéase, de sorte que l'activité de la protéase diminue, voire disparaît, mais ne dénature pas la protéine enzymatique. La sensibilité des différentes protéases aux différentes protéines est différente, il est donc nécessaire d'ajuster la concentration des différentes protéases. Étant donné que la solubilité de l'inhibiteur de protéase dans le liquide est extrêmement faible, il convient de prêter une attention particulière au fait que l'inhibiteur de protéase soit entièrement mélangé dans le tampon afin de réduire la précipitation de l'inhibiteur de protéase. La pepstantine A peut inhiber les protéases acides telles que la pepsine, l'angiotensine, la collagénase, la cathepsine D et la chymosine.

La peptstatine A est un inhibiteur des cathepsines d et e. Après que les cellules HEK293 aient été traitées avec différentes concentrations de pepstatine A pendant 24 heures, l'expression de LC3Ⅱ et de p62 a été détectée. Les résultats ont montré que la pepstatine A pouvait améliorer significativement l’expression de LC3Ⅱ et P62 (P < 0,05) de manière dose-dépendante. 20 µg/ml de pepstatine A ont été utilisés pour traiter HEK293 à différents intervalles de temps, et les effets de différents intervalles de temps sur l'expression de LC3II et de p62 ont été observés. Les résultats ont montré que la pepstatine A pouvait réguler positivement l’expression de LC3II et de p62 en fonction du temps.
Nous sommes un fabricant de polypeptides en Chine, avec plusieurs années d'expérience dans la production de polypeptides. Hangzhou Taijia Biotech Co., Ltd. est un fabricant professionnel de matières premières polypeptidiques, qui peut fournir des dizaines de milliers de matières premières polypeptidiques et peut également être personnalisé en fonction des besoins. La qualité des produits polypeptidiques est excellente et la pureté peut atteindre 98 %, ce qui a été reconnu par les utilisateurs du monde entier. Bienvenue pour nous consulter.

Le cagrilintide est un peptide synthétique qui imite l'action de l'amyline, une hormone sécrétée par le pancréas qui régule la glycémie et l'appétit. Il est composé de 38 acides aminés et contient une liaison disulfure. Le cagrilintide se lie à la fois aux récepteurs de l'amyline (AMYR) et aux récepteurs de la calcitonine (CTR), qui sont des récepteurs couplés aux protéines G exprimés dans divers tissus, tels que le cerveau, le pancréas et les os. En activant ces récepteurs, le cagrilintide peut réduire la prise alimentaire, abaisser la glycémie et augmenter la dépense énergétique. Le cagrilintide a été étudié comme traitement potentiel de l'obésité, un trouble métabolique caractérisé par un excès de graisse corporelle et un risque accru de diabète, de maladies cardiovasculaires et de cancer. Le cagrilintide a montré des résultats prometteurs dans des études animales et des essais cliniques, démontrant une perte de poids significative et un meilleur contrôle glycémique chez les patients obèses avec ou sans diabète de type 2.

Figure 1. Modèle d'homologie du cagrilintide (23) lié à AMY3R. (A) La partie N-terminale de 23 (bleu) est formée par une hélice a amphipathique, profondément enfouie dans le domaine TM d'AMY3R, tandis que la partie C-terminale devrait adopter une conformation étendue qui lie la partie extracellulaire du récepteur. (29,30) L'acide gras attaché à l'extrémité N-terminale de 23, les résidus proline (qui minimisent la fibrillation) et l'amide C-terminal (essentiel à la liaison au récepteur) sont mis en évidence dans les représentations en bâton. AMY3R est formé par CTR (gris) lié à RAMP3 (protéine modifiant l'activité du récepteur 3 ; orange). Le modèle structurel a été créé à l'aide des structures modèles suivantes : une structure complexe de CGRP (récepteur de type récepteur de calcitonine ; code pdb 6E3Y) et une structure cristalline apo du squelette 23 (code pdb 7BG0). (B) Gros plan sur 23 mettant en évidence la liaison disulfure N-terminale, un pont salin interne entre les résidus 14 et 17, un « motif de fermeture éclair à leucine » et une liaison hydrogène interne entre les résidus 4 et 11. (adapté de Kruse T, Hansen JL, Dahl K, Schäffer L, Sensfuss U, Poulsen C, Schlein M, Hansen AMK, Jeppesen CB, Dornonville de la Cour C, Clausen TR, Johansson E, Fulle S, Skyggebjerg RB, Raun K. Développement du Cagrilintide, un analogue de l'amyline à action prolongée. J Med Chem. 12 août 2021;64(15):11183-11194.)
Certaines des applications biologiques du cagrilintide sont :
Le cagrilintide peut moduler l'activité des neurones de l'hypothalamus, la région du cerveau qui contrôle l'appétit et l'équilibre énergétique (Lutz et al., 2015, Front Endocrinol (Lausanne)). Le cagrilintide peut inhiber l'activation des neurones orexigènes, qui stimulent la faim, et activer les neurones anorexigènes, qui suppriment la faim. Par exemple, le cagrilintide peut réduire l'expression du neuropeptide Y (NPY) et du peptide apparenté à l'agouti (AgRP), deux peptides orexigènes puissants, et augmenter l'expression de la proopiomélanocortine (POMC) et du transcrit régulé par la cocaïne et l'amphétamine (CART), deux peptides anorexigènes, dans le noyau arqué de l'hypothalamus (Roth et al., 2018, Physiol Behav). Le cagrilintide peut également renforcer l’effet rassasiant de la leptine, une hormone qui signale l’état énergétique du corps. La leptine est sécrétée par le tissu adipeux et se lie aux récepteurs de la leptine sur les neurones hypothalamiques, inhibant les neurones orexigènes et activant les neurones anorexigènes. Le cagrilintide peut augmenter la sensibilité des récepteurs de la leptine et potentialiser l’activation induite par la leptine du transducteur de signal et de l’activateur de transcription 3 (STAT3), un facteur de transcription qui médie les effets de la leptine sur l’expression des gènes (Lutz et al., 2015, Front Endocrinol (Lausanne)). Ces effets peuvent réduire la consommation alimentaire et augmenter la dépense énergétique, entraînant une perte de poids.

Figure 2. Prise alimentaire chez le rat après administration sous-cutanée de Cagrilintide 23. (adapté de Kruse T, Hansen JL, Dahl K, Schäffer L, Sensfuss U, Poulsen C, Schlein M, Hansen AMK, Jeppesen CB, Dornonville de la Cour C, Clausen TR, Johansson E, Fulle S, Skyggebjerg RB, Raun K. Développement du Cagrilintide, un analogue de l'amyline à action prolongée. J Med Chem. 12 août 2021;64(15):11183-11194.)
Le cagrilintide peut réguler la sécrétion d'insuline et de glucagon, deux hormones qui contrôlent la glycémie. Le cagrilintide peut inhiber la sécrétion de glucagon par les cellules alpha du pancréas, ce qui empêche la production excessive de glucose par le foie. Le glucagon est une hormone qui stimule la dégradation du glycogène et la synthèse du glucose dans le foie, augmentant ainsi la glycémie. Le cagrilintide peut supprimer la sécrétion de glucagon en se liant aux récepteurs de l'amyline et à la calcitonine des cellules alpha, qui sont couplés à des protéines G inhibitrices qui réduisent les niveaux d'adénosine monophosphate cyclique (AMPc) et l'afflux de calcium. Le cagrilintide peut également potentialiser la sécrétion d'insuline par les cellules bêta du pancréas, ce qui améliore l'absorption du glucose par les muscles et le tissu adipeux. L'insuline est une hormone qui favorise le stockage du glucose sous forme de glycogène dans le foie et les muscles, ainsi que la conversion du glucose en acides gras dans le tissu adipeux, abaissant ainsi la glycémie. Le cagrilintide peut améliorer la sécrétion d'insuline en se liant aux récepteurs de l'amyline et à la calcitonine des cellules bêta, qui sont couplés à des protéines G stimulatrices qui augmentent les niveaux d'AMPc et l'afflux de calcium. Ces effets peuvent abaisser la glycémie et améliorer la sensibilité à l'insuline, ce qui peut prévenir ou traiter le diabète de type 2 (Kruse et al., 2021, J Med Chem ; Dehestani et al., 2021, J Obes Metab Syndr.).
Le cagrilintide peut également affecter la fonction des ostéoblastes et des ostéoclastes, deux types de cellules impliquées dans la formation et la résorption osseuse. Les ostéoblastes sont responsables de la production d’une nouvelle matrice osseuse, tandis que les ostéoclastes sont responsables de la destruction de l’ancienne matrice osseuse. L'équilibre entre les ostéoblastes et les ostéoclastes détermine la masse et la résistance osseuse. Le cagrilintide peut stimuler la différenciation et l'activité des ostéoblastes, ce qui augmente la formation osseuse. Le cagrilintide peut se lier aux récepteurs de l'amyline et aux récepteurs de la calcitonine sur les ostéoblastes, qui activent les voies de signalisation intracellulaires qui favorisent la prolifération, la survie et la synthèse matricielle des ostéoblastes (Cornish et al., 1996, Biochem Biophys Res Commun. ). Le cagrilintide peut également augmenter l'expression de l'ostéocalcine, un marqueur de la maturation et de la fonction des ostéoblastes (Cornish et al., 1996, Biochem Biophys Res Commun.). Le cagrilintide peut également inhiber la différenciation et l'activité des ostéoclastes, ce qui diminue la résorption osseuse. Le cagrilintide peut se lier aux récepteurs de l'amyline et à la calcitonine des précurseurs des ostéoclastes, ce qui inhibe leur fusion en ostéoclastes matures (Cornish et al., 2015). Le cagrilintide peut également réduire l'expression de la phosphatase acide tartrate-résistante (TRAP), un marqueur de l'activité des ostéoclastes et de la résorption osseuse (Cornish et al., 2015, Bonekey Rep.). Ces effets peuvent améliorer la densité minérale osseuse et prévenir ou traiter l'ostéoporose, une maladie caractérisée par une faible masse osseuse et un risque accru de fracture (Kruse et al., 2021 ; Dehestani et al., 2021, J Obes Metab Syndr.)
Il convient à l'injection intrathécale et à d'autres méthodes de traitement (telles que les analgésiques systémiques, le traitement adjuvant ou la gaine). Le ziconotide est un inhibiteur calcique puissant, sélectif et réversible de type N, sensible à la tension, qui est efficace pour la douleur réfractaire et ne produit pas de résistance aux médicaments après une administration à long terme, et il ne provoque pas de dépendance physique et mentale, ni de dépression respiratoire potentiellement mortelle due à surdosage. La dose quotidienne recommandée est inférieure, avec un bon effet curatif, une sécurité élevée, moins d'effets indésirables, aucune résistance aux médicaments ni dépendance. Ce produit a d’énormes perspectives de marché en tant qu’analgésique.
Selon des statistiques incomplètes, l'incidence de la douleur dans le monde est actuellement d'environ 35 à 45 % et l'incidence de la douleur chez les personnes âgées est relativement élevée, d'environ 75 à 90 %. Une enquête américaine montre que l'incidence de la migraine est passée de 23,6 millions en 1989 à 28 millions en 2001. Dans une enquête sur la douleur chronique dans six villes de Chine, il a été constaté que l'incidence de la douleur chronique chez les adultes est de 40 % et que le taux de traitement médical est de 35 % ; L'incidence de la douleur chronique chez les personnes âgées est de 65 à 80 % et le taux de consultation d'un médecin est de 85 %. Ces dernières années, les dépenses médicales liées au soulagement de la douleur augmentent d’année en année.
De 2013 à juillet 2015, le Pain Research Center aux États-Unis et plusieurs institutions médicales ont mené une étude observationnelle multicentrique à long terme sur l’injection intrathécale de ziconotide chez 93 patientes adultes de race blanche souffrant de douleurs chroniques sévères. L'échelle de score numérique de la douleur et le score sensoriel global des patients avec injection intrathécale de ziconotide et sans injection de ziconotide ont été comparés. Parmi eux, 51 patients ont utilisé l'injection intrathécale de ziconotide, tandis que 42 patients ne l'ont pas fait. Les scores de douleur de base étaient respectivement de 7,4 et 7,9. La dose recommandée d’injection intrathécale de ziconotide était de 0,5 à 2,4 mcg/jour, qui a été ajustée en fonction de la réponse douloureuse du patient et des effets secondaires. La dose initiale moyenne était de 1,6 mcg/jour, 3,0 mcg/jour à 6 mois et 2,5 à 9 mois. À 12 mois, il était de 1,9 mcg/jour, et après 6 mois, le taux de diminution était de 29,4 %, le taux d'augmentation du contraste était de 6,4 % et le taux d'amélioration du score sensoriel global était respectivement de 69,2 % et 35,7 %. Après 12 mois, le taux de diminution était respectivement de 34,4 % et 3,4 %, et le taux d'amélioration du score sensoriel global était respectivement de 85,7 % et 71,4 %. Les effets secondaires les plus importants étaient les nausées (19,6 % et 7,1 %), les hallucinations (9,8 % et 11,9 %) et les étourdissements (13,7 % et 7,1 %). Les résultats de cette étude ont confirmé une nouvelle fois l’efficacité et la sécurité du ziconotide recommandé en injection intrathécale de première intention.
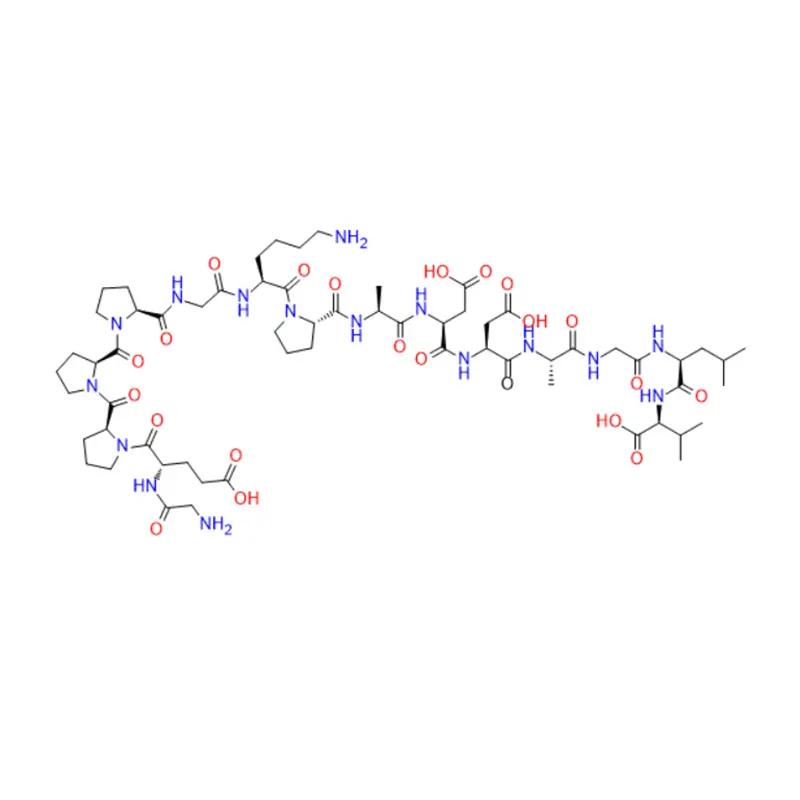
L'étude préliminaire sur le ziconotide remonte aux années 1980, lorsque l'application thérapeutique potentielle de peptides rigides et de type protéine dans le venin du cône a été explorée pour la première fois. Ces conotoxines sont de petits peptides riches en liaisons disulfure, généralement de 10 à 40 résidus de longueur, pour cibler efficacement et sélectivement divers canaux ioniques, GPCR et protéines transporteuses. Le ziconotide est un 25-peptide dérivé de Conus magus, qui contient trois liaisons disulfure, et son court pli β est spatialement disposé en une structure tridimensionnelle unique, ce qui lui permet d'inhiber sélectivement les canaux CaV2.2.
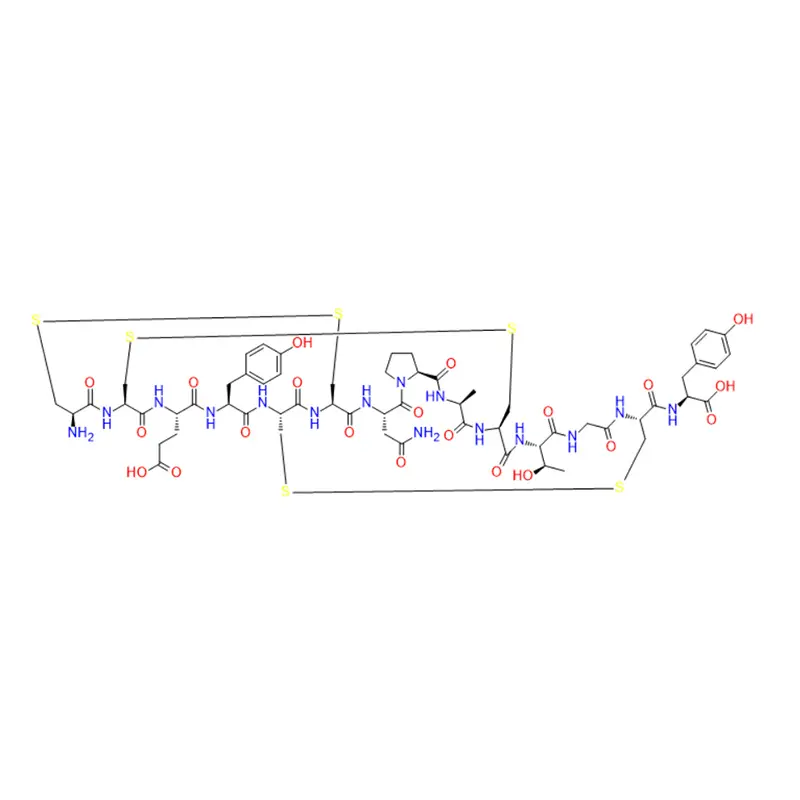
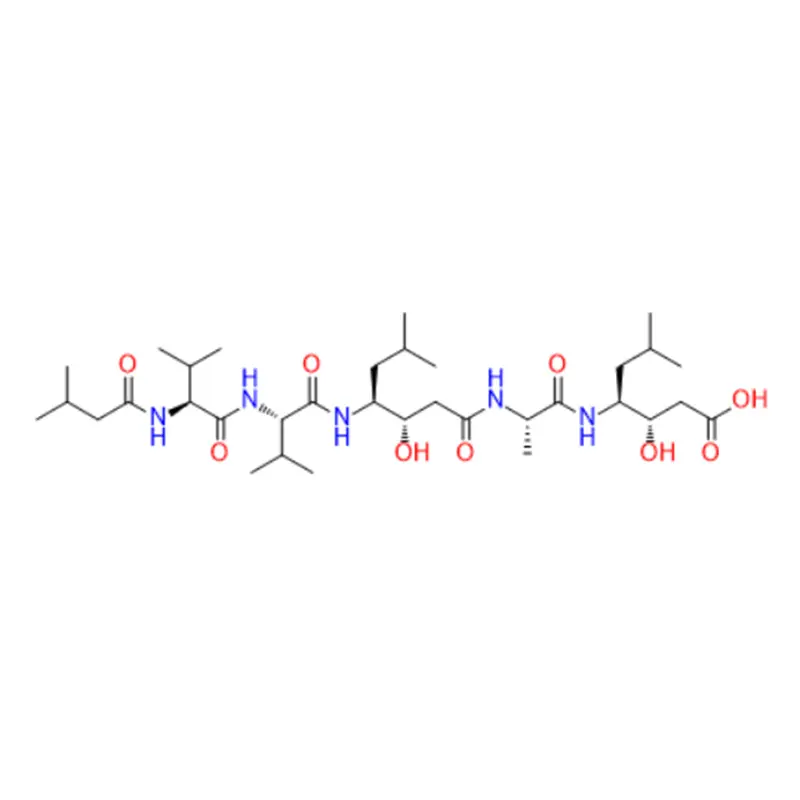
Le linaclotide est un peptide cyclique composé de 14 acides aminés, dont trois sont des cystéines qui forment des liaisons disulfure. Le linaclotide est structurellement apparenté aux peptides endogènes guanyline et uroguanyline, qui sont des ligands naturels du récepteur de la guanylate cyclase C (GC-C). Le récepteur GC-C est exprimé sur la surface luminale des cellules épithéliales intestinales, où il régule la sécrétion de fluides et la motilité intestinale. Le linaclotide se lie au récepteur GC-C avec une affinité et une spécificité élevées et l'active en augmentant les niveaux intracellulaires de guanosine monophosphate cyclique (cGMP). Le GMPc est un deuxième messager qui médie diverses réponses cellulaires, telles que la sécrétion de chlorure et de bicarbonate, la relaxation des muscles lisses et la modulation de la douleur. Le linaclotide agit localement dans le tractus gastro-intestinal et ne pénètre pas la barrière hémato-encéphalique ni n'affecte le système nerveux central. Le linaclotide produit également un métabolite actif, le MM-419447, qui possède des propriétés pharmacologiques similaires à celles du linaclotide. Le linaclotide et son métabolite résistent à la dégradation protéolytique par les enzymes intestinales et sont principalement éliminés sous forme inchangée dans les selles (MacDonald et al., Drugs, 2017).
En activant le récepteur GC-C, le linaclotide augmente la sécrétion de liquide dans la lumière intestinale, ce qui ramollit les selles et facilite les selles. Le linaclotide réduit également l'hypersensibilité viscérale et l'inflammation associées au syndrome du côlon irritable (SCI) et à d'autres troubles gastro-intestinaux. Le linaclotide module l'activité du système nerveux entérique et des nocicepteurs du côlon, qui sont des neurones sensoriels qui transmettent les signaux de douleur de l'intestin au cerveau. Le linaclotide diminue l'expression des gènes liés à la douleur, tels que la substance P et le peptide lié au gène de la calcitonine (CGRP), et augmente l'expression des récepteurs opioïdes, qui assurent l'analgésie. Le linaclotide réduit également la libération de cytokines pro-inflammatoires, telles que l'interleukine-1 bêta (IL-1β) et le facteur de nécrose tumorale alpha (TNF-α), et augmente la libération de cytokines anti-inflammatoires, telles que l'interleukine-10 (IL-10) et le facteur de croissance transformant bêta (TGF-β). Ces effets du linaclotide améliorent les symptômes de constipation et de douleurs abdominales chez les patients atteints du SCI ou de constipation chronique (Lembo et al., The American Journal of Gastroenterology, 2018).
Le linaclotide s'est révélé efficace et bien toléré dans plusieurs essais cliniques impliquant des patients atteints de CC ou d'IBS-C. Dans ces essais, le linaclotide a amélioré les habitudes intestinales, telles que la fréquence, la consistance et l'exhaustivité des selles ; réduction des douleurs et inconforts abdominaux ; et une qualité de vie améliorée et une satisfaction des patients. Le linaclotide a également démontré un profil d'innocuité favorable, la diarrhée étant l'événement indésirable le plus courant. L'incidence de la diarrhée était dose-dépendante et sa gravité était généralement légère à modérée. Les autres événements indésirables étaient généralement similaires à ceux du placebo ou de faible fréquence. Aucun événement indésirable grave ni décès n’a été attribué au traitement par linaclotide (Rao et al., Clinical Gastroenterology and Hepatology, 2015).



Le linaclotide est un médicament nouveau et efficace pour les patients atteints de CC et d'IBS-C qui n'ont pas bien répondu aux thérapies conventionnelles. Il agit en imitant l'action des peptides endogènes qui régulent la fonction et la sensation intestinales. Le linaclotide peut améliorer les habitudes intestinales, réduire les douleurs abdominales et améliorer la qualité de vie de ces patients.

Figure 1. Douleurs abdominales/inconfort abdominal et degré de soulagement du SCI chez les intervenants hebdomadaires au cours des 12 semaines. , placebo ; linaclotide 290 μg.
(Yang, Y., Fang, J., Guo, X., Dai, N., Shen, X., Yang, Y., Sun, J., Bhandari, BR, Reasner, DS, Cronin, JA, Currie, MG, Johnston, JM, Zeng, P., Montreewasuwat, N., Chen, GZ et Lim, S. (2018) Linaclotide dans le côlon irritable syndrome avec constipation : un essai randomisé de phase 3 en Chine et dans d'autres régions. Journal de gastroentérologie et d'hépatologie, 33 : 980-989. est ce que je: 10.1111/jgh.14086.)
Nous sommes un fabricant de polypeptides en Chine, avec plusieurs années d'expérience dans la production de polypeptides. Hangzhou Taijia Biotech Co., Ltd. est un fabricant professionnel de matières premières polypeptidiques, qui peut fournir des dizaines de milliers de matières premières polypeptidiques et peut également être personnalisé en fonction des besoins. La qualité des produits polypeptidiques est excellente et la pureté peut atteindre 98 %, ce qui a été reconnu par les utilisateurs du monde entier. Bienvenue pour nous consulter.
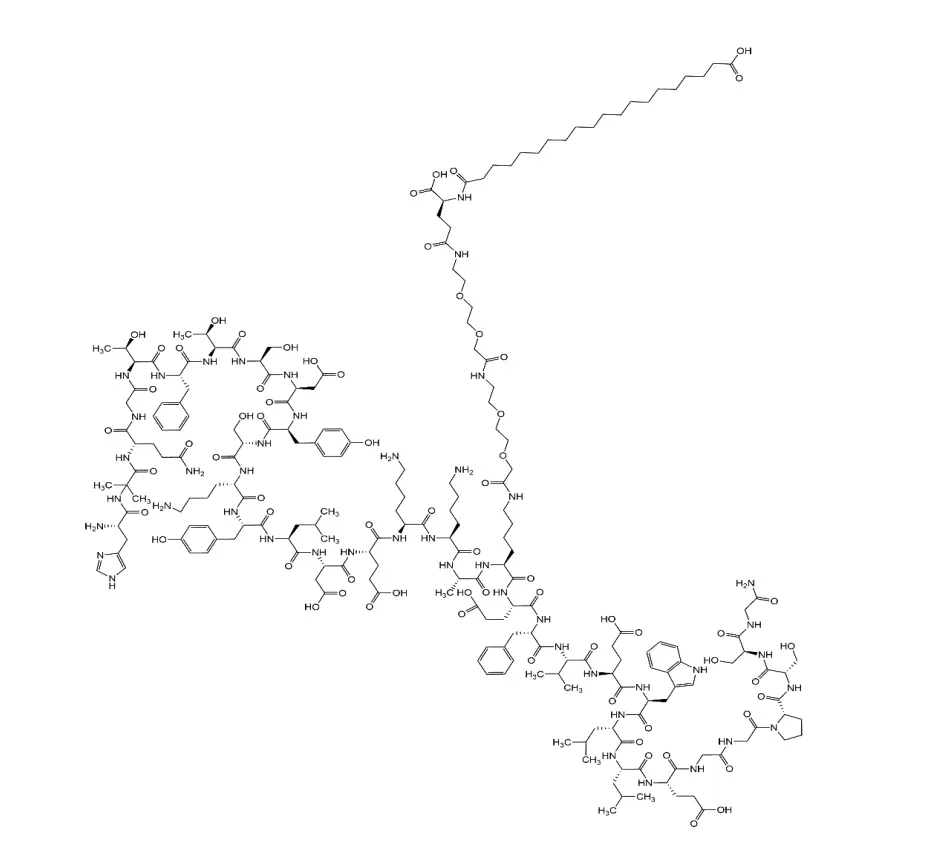



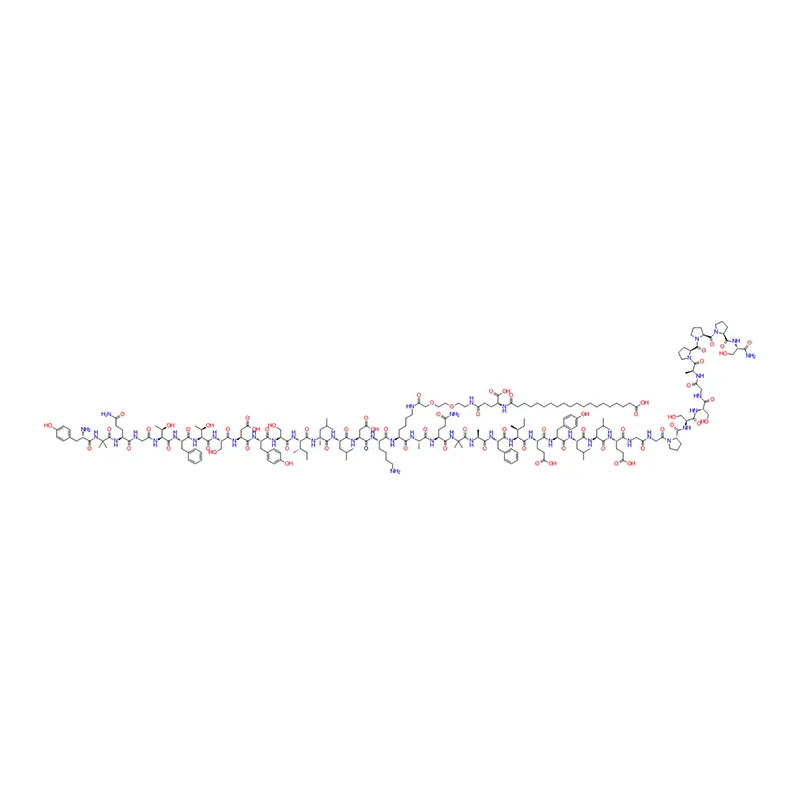
N° CAS : 910463-68-2
Formule moléculaire : C187H291N45O59
Poids moléculaire : 4113,64
Séquence : His-Aib-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-N6-[N-(17-carboxy-1-oxoheptadécyl-L-γ-glutamyl[2-(2-aminoéthoxy)éthoxy] acétyl[2-(2-aminoéthoxy)éthoxy]acétyl]-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Arg-Gly-OH
Application : Un analogue du peptide-1 de type glucagon humain (GLP-1) à action prolongée utilisé pour le traitement du diabète.
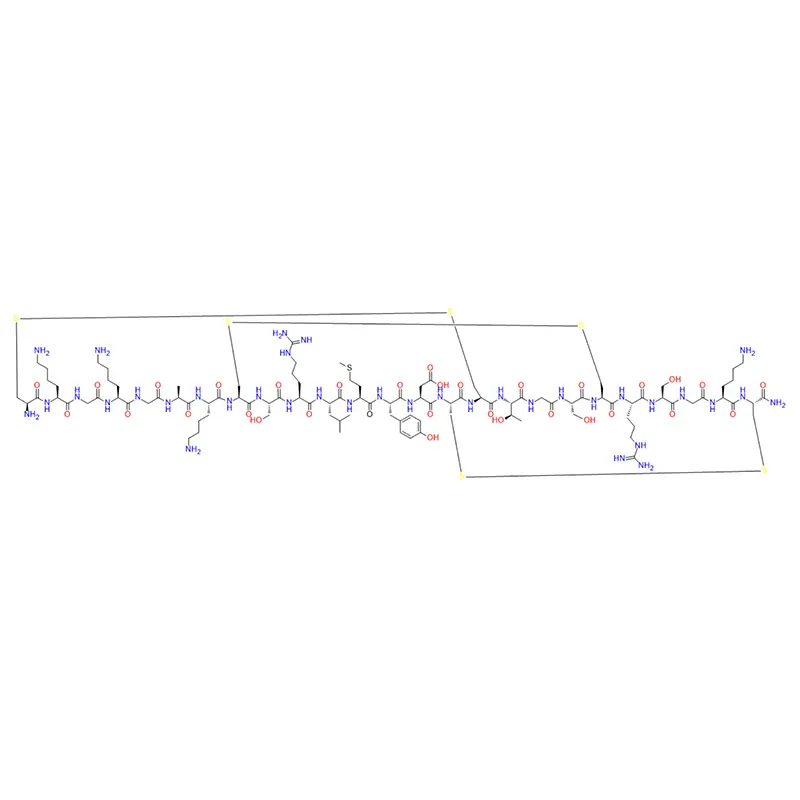
Acétyl hexapeptide -8, également connu sous le nom d'akiréline et d'hexapeptide. L'acétyl hexapeptide -8 est également appelé « ressemblant à la toxine botulique »/« frottis de toxine botulique » par de nombreuses personnes. On peut dire que l’aquiline est un polypeptide antirides ayant un meilleur effet que la toxine botulique.
Comme nous le savons tous, la toxine botulique est un produit de beauté qui nécessite une injection. Son utilisation est extrêmement dangereuse et doit être utilisée par des professionnels. La posologie doit être strictement contrôlée, mais elle ne peut toujours pas éviter divers effets secondaires tels que la raideur faciale et la paralysie faciale.
Argireline a été vérifiée dans les expériences humaines des fabricants de cosmétiques : la profondeur moyenne des rides a diminué de 16,9 % et 27,0 % après 15 et 30 jours avec une solution d'Argireline à 10 %, et le volume des rides a diminué de 20,6 % et la longueur des rides a diminué de 15,9 % après seulement 7 jours avec une solution d'Argireline à 2 %. On constate que l’effet de l’achillerine sur les rides est très significatif.

Les rides de la peau du visage humain sont principalement causées par le relâchement du collagène et la contraction involontaire des muscles. Si la contraction de ces muscles peut être contrôlée, les muscles de la peau peuvent être détendus pour soulager les rides et atteindre l'objectif fondamental d'éliminer les rides.
La toxine botulique, en tant que méthode efficace d’élimination des rides, est largement connue pour son excellent effet. Même si cela entraîne de grands risques après utilisation, il y aura toujours un grand nombre de consommateurs prêts à l'utiliser. Le polypeptide est différent. En tant que produit organique de synthèse, lorsqu’il est utilisé comme ingrédient cosmétique, il peut être rapidement dégradé en acides aminés libres à faible concentration. Sa séquence principale est basée sur le corps humain et son mécanisme d'action est naturel. Les caractéristiques des petits peptides moléculaires leur permettent d'avoir une bonne perméabilité transdermique et d'être bien absorbées par le corps humain. L'acétyl hexapeptide -8 empêche le nerf de transmettre les informations de contraction musculaire via un mécanisme similaire à la toxine botulique, de sorte que le muscle ne puisse pas se contracter pour éliminer les rides. Il a une activité antirides élevée et peu d’effets secondaires et a été largement utilisé dans divers produits cosmétiques haut de gamme.

Nous sommes un fabricant de polypeptides en Chine, avec plusieurs années d'expérience dans la production de polypeptides. Hangzhou Taijia Biotech Co., Ltd. est un fabricant professionnel de matières premières polypeptidiques, qui peut fournir des dizaines de milliers de matières premières polypeptidiques et peut également être personnalisé en fonction des besoins. La qualité des produits polypeptidiques est excellente et la pureté peut atteindre 98 %, ce qui a été reconnu par les utilisateurs du monde entier. Bienvenue pour nous consulter.

Synthèse peptidique en phase solide
Technologie SPPS de pointe pour produire des peptides de haute qualité jusqu'à 150 acides aminés
Synthèse en phase solution
Synthèse personnalisée de peptides complexes nécessitant des méthodologies en phase solution
Modification peptidique
Expertise dans diverses modifications peptidiques, notamment la glycosylation, la PEGylation et la cyclisation
Tests analytiques
Caractérisation complète des peptides par HPLC, MS, RMN et d'autres techniques avancées




À l'heure actuelle, nous pouvons fournir : des glycopeptides, des peptides marqués par des isotopes, des peptides chélateurs macrocycliques, des peptides antigéniques complexes MAPS, qui sont utilisés dans diverses recherches scientifiques ; Toutes sortes de peptides marqués par fluorescence sont appliqués à la détermination de l'activité enzymatique et à l'étude des sondes moléculaires ; Peptide chimique Click, peptide modifié au polyéthylène glycol, peptide cyclique et peptide pénétrant dans les cellules, qui sont appliqués à la recherche de divers médicaments polypeptidiques pour améliorer la demi-vie et l'activité des médicaments polypeptidiques.
Copyright © 2025 Hangzhou Hotide Biotech Co., Ltd. Tous droits réservés.